|
CONNECTIVE TISSUE DISEASES
|
|
|
|
Different types of connective tissue diseases Collagen-vascular diseases: In this group there is damage of connective tissue as a result of complex immunological reactions involving autogenous antigens. This group includes systemic lupus erythematosus, rheumatic fever, systemic sclerosis and dermatomyositis. Genetic group of connective tissue diseases such as in Ehlers-Danlos syndrome. Degenerative group as in scurvy or senile elastosis.
Lupus erythematosus is an auto-immune connective tissue disease. This group includes systemic lupus erythematosus, which is characterized by skin and multisystemic manifestations and discoid lupus erythematous which has only cutaneous manifestations. It has been proposed that genetic factors and somatic mutations be implicated in the pathogenesis of the disease, in which immunoglobulins are present at the dermo-epidermal junction.
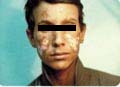
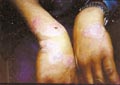
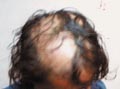
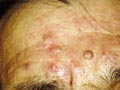
DISCOID LUPUS ERYTHEMATOSUS (DLE) Discoid lupus erythematosus is an auto-immune disease, which is benign but has a very chronic course. The disease affects females more than males, any age group may have the disease but adults are more affected than children. The onset of the lesions may be precipitated by a variety of factors such as mental stress, trauma, infection, excessive exposure to sunlight, PUVA therapy, drugs such as isoniazid and griseofulvin. Clinical Features Localized reddish, scaly, hyperkeratotic, well-defined patches of various-sized, appear on sun exposed areas mainly on face, hands and scalp. The lesions are characterized with follicular plugging known as “stibbling,” adherent scales and horny plugging, which have occupied dilated pilosebaceous canals. Follicular plugging is characteristic. The lesion has a chronic course and tend to heal with atrophy, scarring and pigmentary changes. Lesions of the scalp lead to permanent cicatricial alopecia. Telengectasia is a common manifestation and the lesions usually show hyperpigmentation at the borders.
Diagnosis Discoid lupus erythematosus has a characteristic clinical, laboratory and histopathological features. Histopathology Histopathological changes include five cardinal signs
Laboratory Findings
Differential Diagnosis DLE has to be differentiated from different skin diseases: Systemic lupus erythematosus: DLE may become extensive covering wide areas of the skin simulating systemic lupus but without systemic involvement of internal organs and usually shows negative LE test. Polymorphous light eruption: The lesions are more superficial, scarring is minimal if it happens, have a shorter course than DLE, and the eruption is polymorphous. Seborrheic dermatitis: the sites involved are the seborrheic areas. The skin lesion is sharply demarcated, greasy, scaly patches and heal without scarring or atrophy. Psoriasis: Dry silvery scaly patches showing neither atrophy nor scarring. Treatment Preventive measures are very important. Protecting the patient from direct exposure to sunlight and using sunscreens especially on the seashores. Extreme heat and cold exacerbate the preexisting lesions. Topical or oral steroids or chloroquine helps some. Etretinate.
SYSTEMIC LUPUS ERYTHEMATOSUS (SLE) This is an immune connective tissue disease that has a familial tendency, marked immune dysfunction and characterized by multisystmes involvement. The disease may affect any age group especially adult females. Immunoglobulins, predominantly IgG, but less frequently IgM and IgA, together with complement (C1, C3) can be demonstrated at the dermo-epidermal junction by immunofluorescence techniques. Clinical Features
Fever is usually of the remitting type.
Kidney involvement: hematuria, mild albuminuria, hypertension and edema due to thickening of the glomelular capillaries with focal necrosis known as wire loop lesions. Cardiac changes: Cardiac signs of pericarditis, myocarditis, and endocarditis leading to vegetative lesions of the valves known as “Limban Sacks syndrome“. Vascular changes: may be generalized involving most blood vessels of skin and internal organs leading to different and complex manifestations. Neurological manifestations: Epilepsy and convulsions.
NEONATAL SYSTEMIC LUPUS ERYTHEMATOSUS Neonatal SLE is a rare syndrome occurring in infants of mothers with connective-tissue disease. Clinical Features of Neonatal SLE Skin manifestations The infants have large annular erythematous lesions or discoid lesions that may be present at birth but always develop within the first 2 months, sometimes being precipitated by sunlight. Occasionally, the lesions may be purpuric on both the face and trunk. The rash improves after 4-6 months and completely clears within a year without scarring. Infants with neonatal LE may occasionally develop SLE in the second decade. Neonatal LE can occur in successive pregnancies. It has long been known that the LE-cell factor and antinuclear antibodies could cross the placenta and may be demonstrated in the newborn for as long as 3 months without harming the child. Mothers of babies with congenital heart disease have a one in three chance of developing SLE or other connective-tissue disease. Anti-nuclear factors can be detected in most cases and can be used for prenatal screening. Systemic manifestations Cardiac manifestations as heart block, hepatospleenomegaly, anemia, thrombocytopenia and arthritis, may be associated with the skin lesion. Renal and central nervous system involvement is rare in children but may occur.
SYSTEMIC LUPUS ERYTHEMATOSUS IN CHILDHOOD The clinical picture, course and treatment are similar to the disorder in adults. The earliest age of onset reported is 3 months. Bullous SLE may resemble chronic bullous disease of childhood. Different organs are involved mainly the kidneys showing diffuse proliferatve nephritis, central nervous system manifestations, hepatospleenomegaly and lymph nodes enlargement are common in children, affected by SLE. Laboratory Findings in SLE
TREATMENT OF SYSTEMIC LUPUS ERYTHEMATOSUS In acute cases and during severe exacerbation: Bed-rest is required. Undue exposure to sun: should be avoided and the patients are advised to wear broad-brimmed hats, to cover the ‘V‘ shape area of the neck and the arms and to use a sunscreen preparation. Mental stress: physical overexertion and secondary infection should be avoided. Symptomatic treatment Anemia must be corrected. Drugs: may be required for symptomatic treatment taking care to avoid drugs that may exacerbate the condition, especially antibiotics, which are usually needed to combat infections. Estimation of C-reactive protein may be helpful: a level higher than 60 mg suggests infection. Dapsone: may give some relief for urticarial lesions, bullous eruptions and for thrombocytopenia. Antihypertensive drugs: Hypertension must be treated and it is generally agreed that hydrallazine can be used safely in most patients with SLE. Dieuretics: Nephrotic syndrome or cardiac failures need diuretics. . Low C3 often indicates severe renal disease. Steroids: It should be noted that not all patients require steroids. Some fulminating cases have been treated with massive doses of steroids but the advantages of such therapy rarely outweigh the risks. Prednisone, 60 mg per day, is the steroid of choice initially. Once the condition appears to be under control, the dose may be reduced gradually, until maintenance dose of about 5-15 mg per day is reached. It has been claimed that a single dose daily or on alternate days, before a meal or with milk before bedtime, produces fewer side effects and does not impair the therapeutic response. Prednisone and azathioprine, or prednisone and Cyclophosphamide can give also good results in renal involvement. The erythrocyte sedimentation rate (ESR) is no guide to the adequacy of therapy. The titers of antinuclear antibodies often persist unchanged despite clinical remission. Anti-DNA antibody and serum complement levels may be helpful in predicting exacerbation. Steroid myopathy can occur with high doses. Prolonged high dosage of corticosteroids, for example, 60 mg of prednisone daily for 6 months, is said to improve the renal lesions more than small suppressive doses. This improvement of survival is not seen in patients with raised blood urea before the onset of therapy. There is no evidence that steroids are prophylactic and that prolonged therapy will prevent the development of new features. Serum enzymes are usually normal but urinary creatine is increased Anti-convulsants for epilepsy. Chlorpromazine is a good sedative, for cases accompanied by psychosis. Aspirin: may be very useful, particularly in patients with joint manifestations. There is an increased risk of aspirin hepatotoxicity in SLE. Indomethacin: may improve arthritis. Corticosteroids are required in the acute cases and should be given in adequate dosage. Antimalarial drugs: In mild cases the administration of chloroquine or hydroxychloroquine may allow the dose of steroids to be reduced. Antimalarials are less useful than in DLE and for long-term therapy may be dangerous causing photosensitivity. Pregnancy is not contra-indicated, as healthy live babies have been delivered from women on antimalarial therapy throughout pregnancy. Treatment of cases not responding to prednisolone Adult cases not responding to systemic steroids can be treated by the following medications: Immuno-suppressive drugs: Immunosuppressive drugs have been used for adult patients not responding to corticosteroids but their use should be given to selected cases. Immuno-suppressive drugs are not given to the young age groups having SLE. It has been concluded that azathioprine added nothing to high-dose prednisone treatment in mild or moderate renal disease. Sudden withdrawal may be followed by relapse. Cyclophosphamide may be a more effective immuno-suppressant but it is more toxic than azathioprine. Tripple therapy with prednisone plus azathioprine and Cyclophosphamide had no therapeutic advantage over prednisone and azathioprine. Chlorambucil has been found helpful. Some reports claim excellent results of prednisone combined with Chlorambucil. Methotrexate: this drug is given in a dose of 7.5 mg weekly which has improved steroid-resistant patients. The long-term risk of malignancy must be considered, whenever immunosuppressive drugs are used. Treatment of cases not controlled by prednisolone and immuno-suppressive drugs. Adult cases of systemic lupus erythematous that are not controlled by prednisone and immuno-suppressive drugs, can be put under the following type of treatment: Methyl prednisone:Pulse therapy with 1g methylprednisone is given intravenously in 500 ml normal saline over 4 h on three successive days to inpatients may be helpful. Anticoagulants: may be required for patients with the lupus or anticardiolipin antibodies who have thrombotic episodes. Danazol: 400-600 mg daily may help some patients.
BULLOUS SYSTEMIC LUPUS ERYTHEMATOSUS Vesiculobullous skin eruptions of systemic lupus erythematosus are uncommon. The clinical manifestations are similar to that of bullous pemphigoid and dermatitis herpetiformis. Histologically, the bullae are subepidermal with a neutrophilic infiltrate occasionally resulting in micro-abscesses resembling dermatitis herpetiformis. Immunofluorescence (IF) shows linear bands of IgG, IgA and IgM and C3 in the basement membrane zone.
DISORDERS
CAUSED BY TRANSPLACENTAL TRANSFER OF Clinical manifestations in the neonates have now been reported in a number of maternal disorders that are believed to be induced by circulating autoantibodies. Of particular interest to the dermatologist are those reports concerning lupus erythematosus, pemphigus vulgaris, herpes gestationis and aphthosis. Since IgA, IgM and IgE antibodies do not cross the placenta in significant amounts; this phenomenon is restricted to diseases caused by autoantibodies of IgG class. Complement does not pass across the placenta. Complement can be detected in the fetus from about the 11th week of gestation. Maternal IgG is catabolized more or less completely within the first 3-6 months of life, and antibody-mediated transplacental diseases can be expected to remit spontaneously within this period.
Dermatomyositis is an autoimmune connective tissue disease involving skin and muscles. Children and older age groups may be affected with the disease that may be fatal in some cases. Clinical Features Different skin and muscle manifestations appear in dermatomyositis, which may have vague signs and symptoms making diagnosis more difficult. There are two types of dermatomyositis, the juvenile and the adult type.
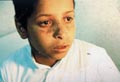
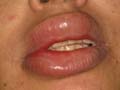
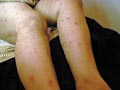
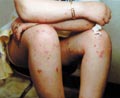
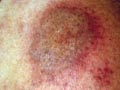
Occurs in childhood, where it has the same manifestation as the adult type with some differences. Raynaud‘s phenomenon is rare in childhood. Muscle involvement is more extensive than the adult type. The prognosis in childhood is better than in adults. Skin manifestations: The characteristic skin manifestations are: Acute cyanotic erythema, erythematous scaly macules or urticarial lesions appear on any site of the skin surface mainly on the face with swollen eyelids. Other skin manifestations mimic those of disseminated lupus erythematosus leading to atrophy and telengectasia; subcutaneous calcification or scleroderma like of the affected skin. In more chronic cases the skin lesions are in the form of: Distinctive lichenoid scaly papules with telengectatic cyanotic atrophy resembling lichen planus, mainly on the back of hands, knees and extensor surfaces of the limbs. These lesions may show pigmentation and calcification. Mucous membrane of the mouth shows aphthous like lesions. Involvement of the scalp leads to cicatricial alopecia. Muscle manifestations: Muscles involved are mainly those of the proximal extremities that become swollen, tender and painful where the inflammatory changes end in contracture deformities. Systemic Manifestations Different systemic manifestations appear according to the muscles involved as dyspnea, dysphagia, diplopia, myocarditis, hepatosplenomegaly and lymphadenopathy. Eye changes: exudates like patches of cotton wool like and hemorrhage appear on opthalmoscopic examination. Laboratory Findings: Hypochromic anemia, leucocytosis and increased sedimentation rate. Serological tests for syphilis are negative in contrast to systemic lupus, which show positive tests. Albuminuria and hematuria L.E test is negative. Tissue enzymes such as aldolase, creatine and phosphokinase are elevated. Differential Diagnosis Systemic lupus erythematosus, scleroderma, erysipelas and myasthenia gravis. Diagnosis Clinical picture: skin, muscle and other organ involvement. Muscle biopsy: usually is diagnostic showing degenerative changes in the muscle with fragmentation, hyalinization with vacuoles and lymphocytic infiltration. Histopathology The connective tissue is thick, condensed and increased in thickness. The collagen shows: edema, homogenization, fibrosis, and sclerosis. The elastic fibers are destroyed. Glandular structure is atrophic. Treatment Treatment of dermatomyositis is usually unsatisfactory. There is high mortality rate. The current available treatments are corticosteroids and immuno-suppressive drugs.
Scleroderma is an autoimmune connective tissue disease characterized by cutaneous thickening due to changes in the dermal connective tissue. The disease is either localized to the skin or systemic, involving the skin and other organs. Types of Scleroderma
Clinical Types:
Guttate morphea: multiple, discrete, sclerotic small lesions occur on the chest and trunk.
Clinical Features The symptoms vary greatly according to the specific organs involved. There is ischaemic atrophy in different skin sites and other systems. Skin manifestations The skin lesions are in the form of large plaques on the chest and limbs having the clinical features of morphea. The lesions may leave fibrotic, pigmented and atrophic areas, while other types may manifest with blistering. Atrophic contracture of the skin and limitation of mobility mainly around the mouth, leading to peri-oral wrinkles, small mouth and difficulty in opening. Atrophic contracture of the eyelids is accompanied by limiatation of the skin mobility, are common manifestations. Telengectasia appears mainly on the face and hands. Generalized pigmentation as that of Addison‘s disease, may accompany some cases. Digital ischaemia leads to micro-infarcts of the finger pulps; loss of finger prints pattern with thromboses and dilated nail folds capillaries, where later there is tapering of the fingers and nail atrophy with ulceration and gangrene. Skeletal manifestations Articular pain, swelling, inflammation of muscles, ankylosis and polyarthritis. Myopathy due to muscle involvement leading to limitations of movement and contracture deformities interfering with mastication. The skin of the face becomes thin and parchment-like, resulting in a peak-like nose and mask face appearance. Osteoporosis, resorption of bones and calcium deposits in the muscles is a common feature. Systemic manifestations Myocardial fibrosis and bundle branch block. Raynaud‘s phenomenon is common. Dyspnea is common and different degrees of lung impairment. Gastro-intestinal: dysphagia and reflux are common, due to limitation of the esophagus mobility, dilatation of the stomach, pyloric stenosis and mal- absorption. Arthritis. Malignant hypertension. Diagnosis Skin manifestations and systemic involvement are usually diagnostic. Raynaud‘s phenomena and sclerodactaly. Increased erythrocyte sedimentation rate. LE Cell is rarely positive Detection of antinuclear factor besides the other manifestations are important data that can help to reach the diagnosis. Biological false positive reaction for syphilis may be positive. Immuno fluorescence technique: thready pattern in the nuclear material may be detected. Complications Ulceration of the skin. Stiffness of the joints. Dyspnea due to affection of respiratory muscles. Death usually occurs from cardiac or acute renal failure. Treatment Treatment is usually unsatisfactory. Symptomatic treatment: Analgesics and anti-rheumatics (Isobrufen) may give relief to joint and muscle pain. General exercise. Hot baths and warmth. High protein diet. Drugs: Thyroid extracts, antihistamines, calcium disodium versenate, and potassium para-amino-benzoic acid are some of the different medications were used for treatment. Different reports concerning the actual value of these medications were reported but some are not convincing. Corticosteroids and immunosuppressive drugs are of limited values. Chloroquine, 250 mg daily for three months may give some improvement.
Lichen sclerosus is a sclerosing immune connective tissue disease, affecting all ages and is associated with auto-antibodies (organ specific antibodies). Clinical Features The primary lesion is polygonal, white slightly raised papule with central depression representing the follicular plugging. Purpura is also a manifestation due to loss of vascular support as a result of degeneration of the dermal collagen. The common sites involved are the back and front of the trunk. Anogenital areas may be involved leading to sclerosis and atrophy of the anal and genital areas. The histologic changes are characteristic. These include liquefaction degeneration of the basal layer, epidermal atrophy, hyperkeratosis and follicular plugging as that of lupus erythematosus.
There is a tendency to local recurrence but, unlike fibrosarcomas, they do not metastasize. Different types of fibromas affecting infants and children are:
Infantile myofibroma is a rare connective tissue disease-affecting infant and may be present at birth and continue to appear for several months. Clinical Manifestations The lesions are due to increased fibrous tissue growth forming nodules in the subcutaneous tissues and bones mainly on the head, neck and trunk. Visceral nodules may affect liver, heart and lungs. The nodules may be rubbery or hard, and may be superficial or deep, invading the muscles. Subcutaneous nodules have good prognosis, but if the viscera are involved, the infant usually dies within few months. Treatment Combination of chemotherapy with Vincristin, Actinomycin D and Cyclophosphamide may give good results.
This begins as a solitary lesion that affects boys much more often than girls. It is present at birth or develops during the first 3 years. The commonest site involved is the axillary and shoulder region, followed by the groin. The lesion presents as a subcutaneous mass, which may enlarge steadily up to a size of 15 cm. Recurrence after excision is infrequent and, if left untreated spontaneous regression may occur.
This is a disorder of glycosaminoglycan synthesis. Clinical Features Skin lesions are present at birth or develop in early childhood. There may be small pearly papules or nodules, particularly on the face or neck. Large subcutaneous tumors may also occur, particularly on the scalp. These may be hard or soft, fixed or mobile, and they may ulcerate. Gingival hypertrophy is commonly present, and flexion contractures of the fingers, elbows, hips and knees may develop. Osteolytic lesions can occur in the skull, long bones or phalanges lead to joint contractures. The musculature is poorly developed. Histological changes show deposition of amorphous hyaline material. The condition persists into adult life that leads to joint contractures and disabling.
|
| Contents | << Previous Chapter | Next Chapter >> | Search |