|
DISEASES OF ABNORMAL KERATINISATION
|
|
|
|
This follicular type begins on the face in infancy. Clinical Manifestations Prominent follicular plugs, which may be filiform, are present from infancy or develop in early childhood on the nose, cheeks and later on the limbs and neck. The keratosis is succeeded by atrophy, which gives rise to atrophoderma on the cheeks. Cicatricial alopecia of scalp and eyebrows starting during childhood or up to the early teenage years is characteristic of the disorder. Some cases may show an associated photophobia, corneal opacity, deafness, nail deformities and mental retardation. There may be numerous milia.
KERATOSIS PILIARIS ATROPHICANS This is a disease of abnormal keratinization that appears at birth or in early infancy. Clinical Features Skin lesions begin usually on the face mainly on the eyebrows as erythematous lesions with small horny plugs that extend to the cheeks and peripherally. Hair follicles may be destroyed leaving areas of alopecia of the eyebrows. Treatment Topical steroid. Keratolytic agent, using low concentration of salicylic acid; 2 per cent in 20 per cent urea topically. Emollient creams.
Acquired icthyosis has the same skin manifestations as the other types but it is caused by different diseases. The syndrome may be associated with other manifestations such as anhidrosis, malabsorption and liver diseases. The diagnosis of an itchy icthysiform skin lesion in infancy or early childhood should exclude the possibility of Hodgkin‘s disease. The different diseases that have icthysiform manifestations besides the other manifestations are: Systemic lupus erythematosus Hodgkin‘s disease. Malignant diseases Nutritional deficiencies Drug reaction as a reaction to medications used for lowering cholesterol.
Pityriasis rubra piliaris (PRP) is a chronic inflammatory disease characterized by fine acuminate, horny, follicular papules that affects both sexes equally and occurs at any age. Etiology The cause of the disease is unknown. The essential abnormality appears to be epidermal over activity. Genetic factors have been considered since the disease is transmitted as an autosomal dominant trait.
JUVENILE PITYRIASIS RUBRA PILIARIS Juvenile pityriasis rubra piliaris of infants and young children may have a different clinical picture than that in older children and adults. The onset of the disease begins usually between the ages of 5 and 10 years. Clinical Manifestations Skin manifestations The eruption usually begins in early childhood as an erythematous slightly scaly macules and papules associated with follicular and perifollicular central keratotic plugging.
The condition is exceedingly chronic followed sometimes by infection. Spontaneous clearing is usually within two years or may progress to generalized exfoliate dermatitis leaving dull, red glazed, atrophic skin that is very sensitive for minor stimuli. The scalp is usually the first site involved that presents with erythema and scaling, leaving cicatricial alopecia after healing of the lesions. Other common sites involved are the sides of the neck, trunk, extensor surfaces of the limbs, the back of the hands, palms and soles, which show hyperkeratosis. The nails may be thickened, dull rough, brittle and become striated in long standing cases. Gooseflesh appearance of the skin due to involvement of extensive area, presenting with sharply marginated patches. Histopathology Hyperkeratosis with keratotic follicular plugs. Parakeratosis, irregular acanthosis, liquefaction degeneration of the basal layer and infiltrate with mast and plasma cells around the dilated blood vessels. Treatment Different lines of treatment are used in the treatment of pityriasis rubra piliaris but most have disappointing results. Topical steroids: alone or in combination with salicylic acid may give some improvement in the dry scaly lesions, especially those involving palms and soles. Systemic steroids: can be used especially in generalized cases with manifestations of erythroderma. Cytotoxic drugs: these drugs should be used under certain strict conditions. These are indicated in older age groups, in reluctant cases and in lesions, which are not responding to other lines of treatments. Retinoids: oral retinoids may give good results. Isotretinoin may improve some cases. Vitamin A In high doses is believed to give encouraging results, but care of hypervitaminosis. A which may elicit a skin eruption. PUVA: these may carry hazardous side effects, especially in infants. The results may be unencouraging. We prefer that this line of treatment should not be used and not recommended in young age group.
DARIER‘S
DISEASE This is a genetic disorder of abnormal keratinization which is characterized by persistent eruption of hyperkeratotic papules mainly on the seborrheic areas. Clinical Features The lesions appear mainly on the seborrheic areas of the face, scalp margins, forehead, ears, nasio-labial folds, upper chest and back. Darrier‘s disease presents with several clinical types:
The primary skin lesions are firm greasy, crusted papules, skin colored, yellowish or brown that may be misdiagnosed as acne or seborrheic dermatitis. If the adherent crust is removed, a central funnel-shaped orifice may sometimes be exposed. Scalp lesions: show heavy crusted papules like seborrhea, but has a characteristic spiny feel to palpation. Flexural lesions: appear mainly on the anogenital region, the groins and the natal cleft. Coalescence of the papules produces irregular warty plaques or papillomatous masses, which in the flexures become vegetative and malodorous. Flexural lesions may simulate seborrheic dermatitis, fungal infections or psoriasis of the flexural areas. The palms and soles: may show hemorrhagic macules and punctate keratoses or minute pits. Nail changes: are characteristics either showing white or red longitudinal bands or V shaped red and white bands at the free margin of the nail. Mucous membranes lesions: are white umbulicated or “cobblestone“ papules on the palate, tongue, buccal mucosa, epiglottis, and pharyngeal wall simulating leucoplakia vulva, esophagus or rectum. Viral and pyogenic infections: Patients with Darrier‘s disease appear to have an increased susceptibility to herpes simplex infections, Pox virus infection Kaposi‘s varicelliform eruption and increased incidence of chronic pyogenic infection. Histopathology The histology shows a distinctive form of dyskeratosis, with corps ronds and supra basal acantholysis. Treatment Many patients with mild disease require no treatment other than a simple emollient and instructed to expose affected areas to sun. For those with more severe disease, the use of etretinate or isotretinoin usually results in significant improvement. Etretinate may give good results. The dose is 0.5-1.0 mg/kg/d. Isotretinoin 1-2 mg/kg/d is another medication that can help some cases. Resurfacing by CO2 Laser and Dermabrasion can be used for severe warty cases especially in intertriginous hypertrophic types.
Acro-keratosis verruciformis is a disease of abnormal keratinization inherited as an autosomal dominant, but sporadic cases also occur. The eruption affects both sexes and is usually present at birth or appears in early childhood. Clinical Features Skin-colored warty papules, which are flat or convex, present on the back of the hands, feet, knees, elbows, and on the forearms. Small groups or isolated papules may develop in other sites. The palms, which may be diffusely thickened, show small keratoses and punctiform breaks in the dermatoglyphic pattern. Friction of the lesions may cause blister formation. The nails may be thickened and white.
DIFFUSE
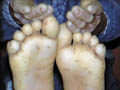
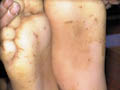
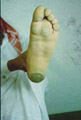
PALMOPLANTAR KERATODERMA The skin lesions may appear in the first few months of life and is usually obvious by the age of four. Clinical Features Skin lesions are even, very thick, yellow hyperkeratotic patches that involve the whole foot, starting on the heel and anterior arch, spreading later to the palms causing thickening of nails. Marked hyperhidrosis is common and usually predispose to fungal infections.
PROGRESSIVE
PALMOPLANTAR KERATODERMA This is a rare syndrome that runs in some families having marriages from their relatives. Clinical Features Skin manifestations The keratoderma is characterized by extension onto the dorsal surfaces of the hands and feet and over the knees and elbows. The condition is associated with eczema that is often secondarily infected. In early infancy, scaling and thickening soon follow redness of palms and soles. This is usually diffuse but sometimes appear in islands, extending to the dorsal surfaces in a glove-like distribution. General Manifestations Hyperhidrosis. Nail thickening or koilonychia,
Fluro-uracil ointment may give temporary improvement. Retinoids may be of some value.
PAPILLON-LE
FEVRE SYNDROME This is an inherited disease of abnormal keratinization affecting infants and young children. Clinical Features Skin manifestations Redness and thickening of the palms and soles simulating psoriasis. General manifestations Hyperhidrosis that may cause an unpleasant odor. Hair is usually normal but may be sparse. Frequent pyogenic infections. Periodontosis resulting in severe gingivitis that may predispose to loss of the teeth . The permanent teeth may be lost in the same fashion. Dural calcification, especially in the attachment of the tentorium and choroid, has been noted in some cases.
Callosities represent variants of abnormal keratinization, which are more common in adults. The condition may be congenital or acquired. Callosities may appear early in young age as a familial type or may be acquired in response to repeated trauma or friction as by tight shoes over the bony prominence of the palms or soles. The most common site is over the head of the third metatarsals where the lesion may be mistaken as skin wart.
Corns may show a central degenerative core in the middle of dense hyperkeratosis, which should be distinguished from the black thrombotic vessels of the verruca. Treatment Mild callosities respond to local application of salicylic acid (20%) and lactic acid (20%) in collodion base for few days. Before each application the dead tissues can be removed by shaving. Extensive cases can be resurfaced by CO2 laser.
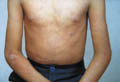
Acanthosis nigricans is an inherited genodermatosis that is characterized by hyperkeratosis and skin pigmentation. The affected skin is covered by papillomatous elevations, which give it a velvety texture. Clinical Features There are different clinical types of acanthosis nigricans:
There is skin thickening and the skin lines are further accentuated. The surface becomes mammillated or rugose, and larger warty excrescencies develop. The intertriginous areas show warty lesions where the lesions may become generalized and cover an extensive areas of the skin surface.
KERATODERMAS DUE TO OTHER DERMATOSES Different skin diseases may give rise to palmo planter hyperkeratosis. These include:
This disease of abnormal keratinization may be familial which is inherited as an autosomal dominant disorder or an acquired in the course of certain diseases. Clinical Features The lesions may be single or multiple appearing as annular, dry plaques surrounded by a raised, fine keratotic wall and sometimes show furrow on the surface. The center is often atrophic but may be hyperkeratotic. The most common sites involved are the limbs, the face and genitalia that show a tendency of centrifugal spread. Mucous membranes may be affected as that of oral mucosa and cornea. Types of Porokeratosis The linear or zosteriform: Linear lesions affect mainly the limbs. Linear porokeratosis may be found together with disseminated actinic porokeratosis. Disseminated superficial actinic Porokeratosis: This is a very common variety occurs in middle-aged individuals who have a history of over exposure to sun where the lesions are mainly on the sun exposed sites without malignant changes. Treatment Keratolytics: such as 2 per cent salicylic acid ointment. 5% 5-fluorouracil ointment is also an effective medication. Cryotherapy. Carbon dioxide laser can be used to extensive lesions. Etretinate is another line of treatment but side effects as erythema and irritation may sometimes limit its use.
|



| Contents | << Previous Chapter | Next Chapter >> | Search |